ecalj MainDocument
This is MainDocument of ecaljdoc. All files are linked from this file.
⚠️ TOML migration (2026-05) — Fortran binaries now read
ctrlg.<sname>.toml+PB.<sname>.tomlonly. Examples below referring toctrl.<sname>/GWinputare legacy; convert withLegacy2toml.py <sname>before running. See TOML migration for the full guide, and migrated Samples/ (EPS, PROCAR, MLOsamples, TestInstall) as templates.
- Here we give GetStarted, together with install and overview of QSGW.
- We have UsageDetails in another file.
- Qiita Japanese may be a help, but most of all are shown here.
- When link did not work, go to https://github.com/tkotani/ecalj to download.
Licence
AGPLv3. For publications, we hope to make a citation as; [1] ecalj package available from https://github.com/tkotani/ecalj/.
AI agent
We use Claude Code to install and use ecalj. For cluster workflows (qsub / sbatch / bash launchers), Claude Code handles job submission and monitors the running jobs (tail logs, react to errors, restart on failure, etc.) without you having to babysit the queue.
The TOML-aware doc and tooling — ctrlgenToml.py, Legacy2toml.py, testecalj, the ctrlg.<sname>.toml schema annotations, much of ecalj_auto's slot scheduler — are co-developed with Claude Code in the same way; many of the doc pages here also carry an explicit "auto-edited by Claude" disclosure where appropriate.
Install
To install ecalj, look into install, as well as install for ISSP.
Features of ecalj package
All electron full-potential PMT method The PMT method means; a mixed basis method of two kinds of augmented waves, that is, APW+MTO. In other words, the PMT method= the linearized (APW+MTO) method, which is unique except the Questaal having the same origin with ecalj. We found that MTOs and APWs are very comlementary, corresponding to the localized and the extented natures of eigenfunctions. That is, very localized MTOs (damping factor where $\kappa \sim 1 $ bohr; this implies only reaching to nearest atoms) together with APWs (cutoff is Ry) works well to get reasonable convergences. We can perform atomic-position relaxation at GGA/LDA level. Because of including APWs, we can describe the scattering states very well.
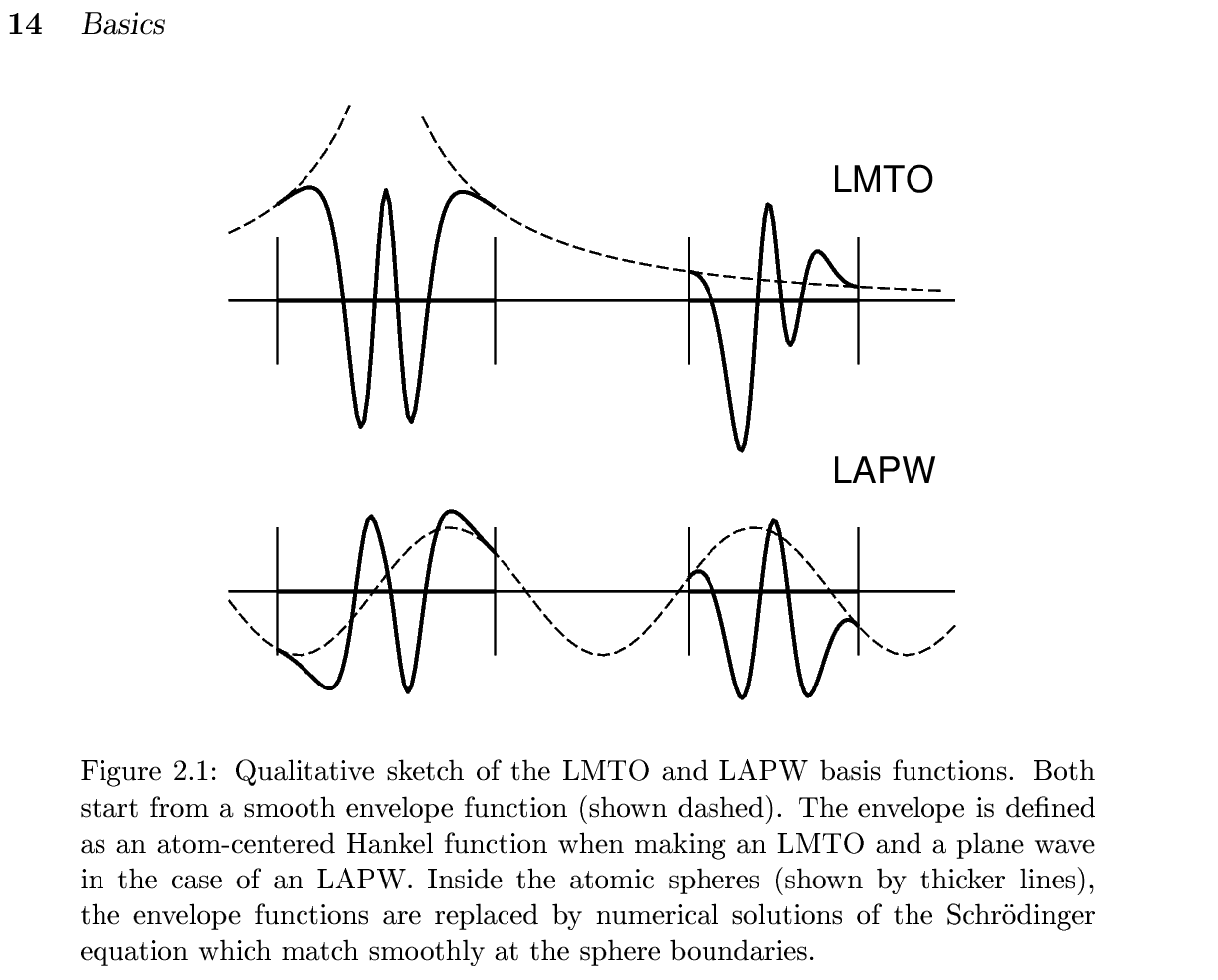 (This fig is taken from nfp-manual by M. Methfessel and M. van Schilfgaarde) Our current PMT formulation is given in
(This fig is taken from nfp-manual by M. Methfessel and M. van Schilfgaarde) Our current PMT formulation is given in[1]KotaniKinoAkai2015, PMT formalism
[2]KotaniKino2013, PMT applied to diatomic molecules.
Since we have automatic settings for basis-set parameters, we don't need to be bothered with the parameter settings. Just crystal structures (POSCAR) are needed for calculations.
- Original PMT was started at https://journals.aps.org/prb/abstract/10.1103/PhysRevB.81.125117
- PMT uses smooth Hankel functions described in the smooth Hankel paper by E. Bott, M. Methfessel, W. Krabs, and P. C. Schmidt, which was used in [B]nfp paper by M. Methfessel and M. van Schilfgaarde, and R.A. Casali. Our PMT is on top them.
- In addition to PMT basis, we use local orbitals together.
- The cutoff Ry may sound surprisingly small. However, note that high energy part of the plane wave methods are consumed only for the core like shape of eigenfunctions with negative curvatures of radial functions. Furthermore, the plane waves have difficulties not satisfying the periodic gauge https://arxiv.org/pdf/2010.03959. On the other hand, localized basis methods such as LMTO and Gaussians have difficulties to handle vacuum regions.
PMT-QSGW method
The PMT-QSGW means
the Quasiparticle self-consistent GW method (QSGW) based on the PMT method. The development of [3] is the key to perform QSGW on the PMT. We can handle even magnetic metals. Since we have implemented ecalj on GPU, we can handle ~40 atoms with four GPUs. [3]Kotani2014, Formulation of PMT-QSGW method [4]D.Deguchi PMT-QSGW applied to a variety of insulators/semiconductors [5]M.Obata GPU implementation, where we treat Type II GaSb/InAs (40 atoms) with four GPUs. We can easily make band plots without the Wanneir kinds of interpolations. This is because an interpolation scheme of self-energy is internally built in. This is very essential to calculate physical quantities from eigenfunctions on top of the QSGW ground states.Since we treat all the electrons, we are not bothered with the ambiguities of the pseudopotentials.
Dielectric functions and magnetic susceptibilities We can calculate GW-related quantities such as dielectric functions, spectrum function of the Green's functions, Magnetic fluctuation, and so on. Since our QSGW can generate eigenfunctions and eigenvalues at any k points.
The Model Hamiltonian with Wannier functions We can generate the effective model (Maxloc Wannier and effective interaction between Wannier funcitons). This is originally from codes by Dr.Miyake, Dr.Sakuma, and Dr.Kino. The cRPA given by Juelich group is implemented. Here is a latest paper to using this, https://journals.aps.org/prb/abstract/10.1103/PhysRevB.108.035141.
- We are now replacing MLWF with MLO (MuffinTinOrbail-based Localized Orbitals), where we can easily throw away (downfolding) the APW degree of freedom.
Singleton pattern in fortran The main part of our codes is in fortran. Our codes have the object-oriented structures respecting the singlton design pattern. We think this is a step to replace our fortran codes with those in modern languages such as JAX/PyTorch. Inevitably, we still have legacy codes but encapsulated as possible, and going to be replaced.
Overview of QSGW
- Band calculations (LDA level) are performed with the program
lmf. The initial setting file isctrlg.<sname>.toml(<sname>is the user-chosen material extension; legacy:ctrl.<sname>). Before runninglmf, it is necessary to runlmfa, which is a spherically symmetric atom calculation to determine the initial conditions for the electron density (lmfafinishes instantaneously). - A file
sigm.<sname>is the key for QSGW calculations. The filesigm.<sname>contains the non-local potential . By adding this potential term to the usual LDA calculation performed bylmf, we can perform QSGW calculations. See figure below. - Thus the problem is how to generate . This is calculated from the self-energy , which is calculated in the GW approximation. Roughly speaking, we obtain with removing the omega-dependence in .
- Therefore, the calculation of is the major part of the QSGW cycle, and is calculated in a double-structure loop. That is, there is an inner loop of
lmf, and an outer loop to calculates using the eigenfunctions given bylmf. This outer loop can be executed with a python script called gwsc (which runs fortran programs). The computational time for QSGW is much longer than that of LDA calculation. As a rule of thumb, it takes about 10 hours for 20 atoms (depending on the number of electrons. For systems more than 10 atoms per cell or so, we recommend to use GPUs). - Here is the QSGW cycle shown in Figure 1 in https://arxiv.org/abs/2506.03477 . MPB meand the mixed product basis to expand products of eigenfunctions.
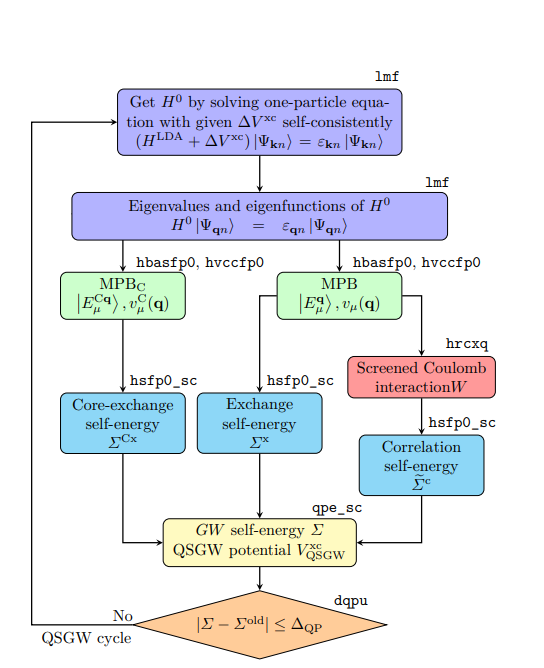
- We have GPU acceleration for QSGW, which also describe basics of QSGW. QSGW algorism fits to GPU computations very well. With four GPUs, we can compute systems with 40 atoms per cell. (As for lmf part, GPUs are not efficiently used yet.). Our PMT allows us to handle large vaccum region for slab model.
- We can perform QSGW virtually without parameter settings by hands. Thus I think ecalj is one of the easiest to perform GW/QSGW. See band database in QSGW at https://github.com/tkotani/DOSnpSupplement/blob/main/bandpng.md (this is a supplement of https://arxiv.org/abs/2507.19189). This is away from complete database, but showing the ability of ecalj.
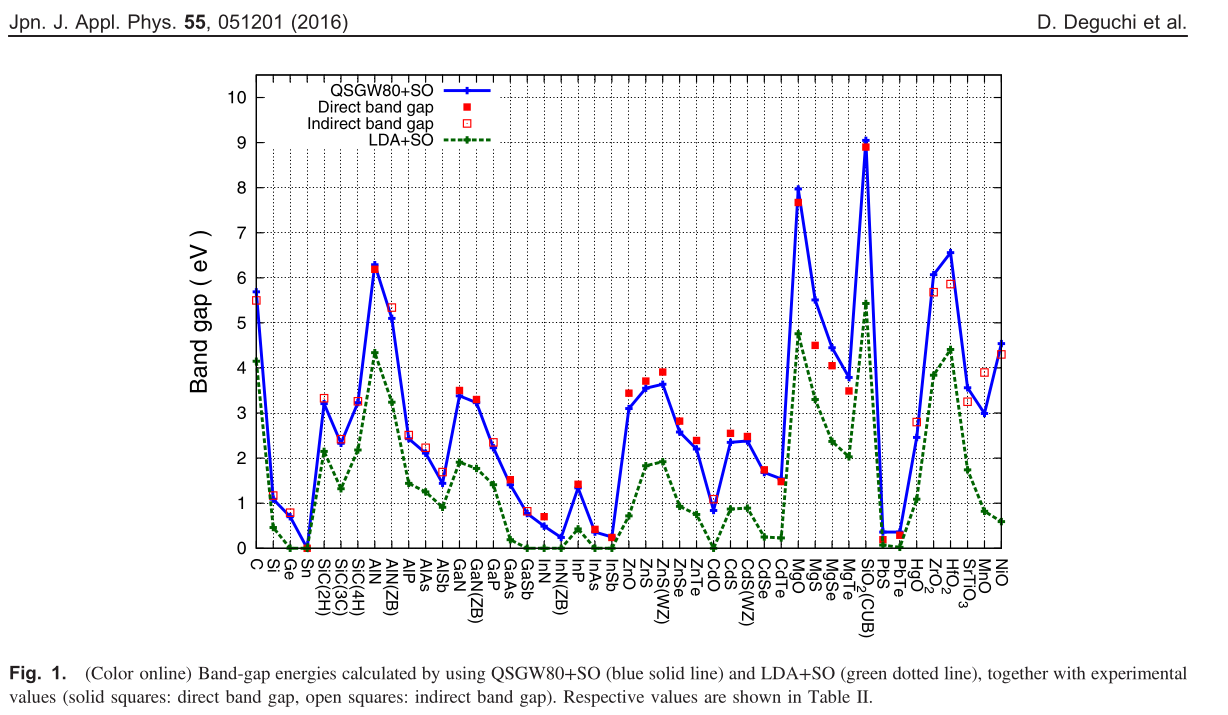 This is taken from [4]D.Deguchi
This is taken from [4]D.Deguchi
In comparison with LDA, we see differences in QSGW;
- Band gap. QSGW tends to give slightly larger than experiments. It looks systematic as in the Figure above.
- Band width. Usually, sp bands are enlarged (except very low density case such as Na). This is consistent with the case for homogeneous electron gas. Localized bands like 3d electrons get narrowed.
- Relative position of bands. e.g. O(2p) v.s. Ni(3d). More localized bands tends to get more deeper. Exchange splitting between up and down get larger (like LDA+U) . In cases such as NiO, magnetic moment become larger; closer to experimental values.
- Hybridization of 3d bands with others. QSGW tends to make eigenfunctions localized.
However, reality is complexed, and not so simple in cases.
GetStarted
Here we explain DFT/QSGW calculations with ecalj. Then we explain how to make band plots. For simplicity, we treat paramagetic cases (nsp=1), no 4f, no SOC. We explain things step by step.
Worked example — every step below uses GaAs as the running example. A minimal seed (
ctrls.gaas+ the generatedctrlg.gaas.toml+PB.<sname>.toml) lives atSamples/GetStarted/GaAs/. Copy that directory to a fresh work area and follow Steps 1-6 below. See its README for the one-shot reproduction script.
Further details are explained at UsageDetailed
Step 0. Get POSCAR
We first need POSCAR (crystal structure in VASP format). You can find samples of POSCAR in ecalj/ecalj_auto/INPUT/testSGA/POSCARALL as
cd ecalj
mkdir TEST
cd TEST
mkdir test1
mkdir test2
cat ecalj_auto/INPUT/testSGA/joblist.bk
cp ../ecalj_auto/INPUT/testSGA/POSCARALL/POSCAR.mp-2534 test1
cp ../ecalj_auto/INPUT/testSGA/POSCARALL/POSCAR.mp-8062 test2For example, POSCAR of mp-2534 GaAs is given as:
Ga1 As1
1.0
3.5212530000000002 0.0000000000000000 2.0329969999999999
1.1737510000000000 3.3198690000000002 2.0329969999999999
0.0000000000000000 0.0000000000000000 4.0659929999999997
Ga As
1 1
direct
0.0000000000000000 0.0000000000000000 0.0000000000000000 Ga
0.2500000000000000 0.2500000000000000 0.2500000000000000 AsThis is POSCAR for ba2pdo2cl2 (QSGW results are shown below):
POSCAR_ba2pdo2cl2
1.0
-2.06443 2.06443 8.40383
2.06443 -2.06443 8.40383
2.06443 2.06443 -8.40383
Ba Pd O Cl
2 1 2 2
Cartesian
0.0 0.0 6.5153213224
0.0 0.0 10.2923386776
0.0 0.0 0.0
0.0 2.06443 0.0
2.06443 0.0 0.0
0.0 0.0 3.1625293056
0.0 0.0 13.6451306944If you have cif and like to convert it to POSCAR, do cif2cell foobar.cif -p vasp --vasp-cartesian --vasp-format=5.
Step 1. convert POSCAR to ctrls
Then we convert POSCAR to ctrls by vasp2ctrl. ctrls is the structure file used in ecalj.
vasp2ctrl POSCAR.mp-2534
mv ctrls.POSCAR.mp-2534.vasp2ctrl ctrls.POSCAR.mp-2534
cat ctrls.mp-2534ctrls.mp-2534 contains crystal structure equivalent to POSCAR:
STRUC
ALAT=1.8897268777743552
PLAT= 3.52125300000 0.00000000000 2.03299700000
1.17375100000 3.31986900000 2.03299700000
0.00000000000 0.00000000000 4.06599300000
SITE
ATOM=Ga POS= 0.00000000000 0.00000000000 0.00000000000
ATOM=As POS= 1.17375100000 0.82996725000 2.03299675000Indentation is needed to show blocks of categories, STRUC and SITE. Another example of ctrls.gaas (the worked-example seed shipped at Samples/GetStarted/GaAs/ctrls.gaas):
%const bohr=0.529177 a=5.65325/bohr
STRUC
ALAT={a}
PLAT=0 0.5 0.5 0.5 0 0.5 0.5 0.5 0
SITE
ATOM=Ga POS=0.0 0.0 0.0
ATOM=As POS=0.25 0.25 0.25ctrls.srtio3:
%const au=0.529177
%const a0=2*1.95/au v=a0^3 a1=v^(1/3)
STRUC ALAT={a1}
PLAT=1 0 0 0 1 0 0 0 1
SITE
ATOM=Sr POS=1/2 1/2 1/2
ATOM=Ti POS= 0 0 0
ATOM=O POS=1/2 0 0
ATOM=O POS= 0 1/2 0
ATOM=O POS= 0 0 1/2%const lines define run-time variables expanded by ctrlgenToml.py (and the legacy ctrlgenM1.py). Multiple definitions on one line are fine, math operators (+ - * / ** ^) and sin / cos / log / sqrt / pi are evaluated, and earlier defines are visible to later ones (so a0=2*1.95/au works because au was defined on the previous %const line). 1/3 is float division (Python 3), so v^(1/3) ≡ v**(1/3) is the cube root.
Gotcha — do not use empty-value placeholders like
%const x= y=.... An entry with no right-hand side merges with the next token and silently breaks the rest of the line (soy,z, ... never get defined). Either drop the empty entry or assign a real value (%const x=0 y=...).
The default names of atomic species are shown by ctrlgenToml.py --showatomlist. Instead of such default symbols, we can use your own symbol as
SITE
ATOM=M1 POS=1/2 1/2 1/2
ATOM=M2 POS= 0 0 0
ATOM=O POS=1/2 0 0
ATOM=O POS= 0 1/2 0
ATOM=O POS= 0 0 1/2
SPEC
ATOM=M1 Z=38
ATOM=M2 Z=22
ATOM=O Z=8, where we need SPEC. Thus AF magnetic NiO is given as
%const bohr=0.529177 a=7.88
STRUC
ALAT={a} PLAT= 0.5 0.5 1.0 0.5 1.0 0.5 1.0 0.5 0.5
SITE
ATOM=Niup POS= .0 .0 .0
ATOM=Nidn POS= 1.0 1.0 1.0
ATOM=O POS= .5 .5 .5
ATOM=O POS= 1.5 1.5 1.5
SPEC
ATOM=Niup Z=28 MMOM=0 0 1.2 0
ATOM=Nidn Z=28 MMOM=0 0 -1.2 0
ATOM=O Z=8 MMOM=0 0 0 0. If you like to use antiferro symmetry (only calculate up band only, and generate down band). See AFsymmetry. MMOM is the initial condition of magnetic moments at sites. The numbers (here 1.2) can be not so accurate (just integers or so). Samples are in minidatabase. Simple materials first. But it is not so difficult to reproduce results by VASP or in MP from your POSCAR file.
- MEMO:
- ctrl2vasp ctrl.mp-2534 can convert back to VASP file. Check this by VESTA. We can use viewvesta (convert and invoke VESTA).
- many unused files are generated (forget them).
- you can use any name for sites such as Niup or something, in such a case you have to set SPEC section in addition. Non integer number of Z is allowed.. Learn afterward.
- In old ctrls, you may see NL,NBAS,NSPEC, which are not necessary now.
Step 2. Get ctrlg.<sname>.toml from ctrls
ctrlg.<sname>.toml plus PB.<sname>.toml is the input pair the Fortran binaries (lmf, lmfa, lmchk, gwsc, ...) actually read since 2026-05. PB.<sname>.toml, which we do not edit usually, is for product basis setting only in GW — auto-emitted by the generator below; hand edits live in ctrlg.<sname>.toml. Generate both files straight from the lightweight ctrls.<sname> (the structure-only seed produced by vasp2ctrl in Step 1) with ctrlgenToml.py:
ctrlgenToml.py mp-2534(Or, for the GaAs worked example shipped at Samples/GetStarted/GaAs/: ctrlgenToml.py gaas — produces the ctrlg.gaas.toml
PB.gaas.tomlthat ship in that directory.)
This single command:
- fills top-level (
symgrp/verbose/time) and[struc] / [[site]] / [[spec]]from the periodic-table defaults (the sameatomlisttable thatctrlgenM1.pyuses); - internally runs
lmfa → lmf --jobgw=0 → gwinitto populate[gw] / [product_basis] / [blocks]and the per-atom tables inPB.<sname>.toml; - writes
ctrlg.<sname>.tomlandPB.<sname>.tomland stops.
Successful end-of-run looks like:
ctrlgenToml: wrote ctrlg.mp-2534.toml (2 spec, 2 sites)
ctrlgenToml: running lmfa -> lmf --jobgw=0 -> gwinit to fill GW sections
ctrlgenToml: done. ctrlg.mp-2534.toml has top-level/[struc]/[[site]]/[[spec]]/...
plus [gw]/[product_basis]/[blocks]. PB.<sname>.toml has nlx/valence/core.Recommended workflow: run ctrlgenToml.py <sname> with no other flags to get the defaults, then edit ctrlg.<sname>.toml afterwards — the file is fully commented and TOML-typed, so in-place edits in your editor are the natural path. The CLI flags listed by ctrlgenToml.py --help are bake-in equivalents of those edits, not requirements. They are useful when scripting / batch- generating many materials, but for normal interactive use leave them off and edit the generated ctrlg.<sname>.toml keys directly.
PB.<sname>.toml is consumed only on the GW path and does not normally need hand editing; leave it as generated.
Pure DFT / no-GW directory? Add --skipgw:
ctrlgenToml.py <sname> --skipgw # ctrlg.<sname>.toml without [gw]/[product_basis]/[blocks];
# no PB.<sname>.toml written.To add the GW sections later without losing the ctrl-side edits you have made in the meantime, use --addgw:
ctrlgenToml.py <sname> --addgw # appends [gw]/[product_basis]/[blocks]
# and writes PB.<sname>.toml in place;
# ctrl-side keys ([bz], [ham], [[spec]], ...)
# are preserved verbatim.--addgw refuses to run if [gw] is already present (to avoid silent duplication). Do not plain re-run ctrlgenToml.py <sname> for this purpose: the default flow overwrites the whole ctrlg.<sname>.toml from ctrls.<sname> (the previous file is moved to ctrlg.<sname>.toml.bakup, one-generation backup only — your edits would be in there but you'd have to merge them back by hand).
Common edits in ctrlg.<sname>.toml:
[bz].nkabc— k-mesh (LDA).[ham].nspin— 2 for magnetic, 1 nonmagnetic.[ham].so— 0 / 1 (full L·S) / 2 (Lz·Sz only); needsnspin = 2.so=1does not yet support QSGW; for QSGW + SOC, run withso=0orso=2to obtainsigm.<sname>, then setso=1.[ham].xcfun— 1 = VWN, 2 = Barth-Hedin (default), 103 = PBE-GGA.[ham].scaledsigma— QSGW mixing.1.0= full QSGW,0.8= QSGW80.(scaledsigma) × (V_xc^QSGW − V_xc^LDA)is added to the lmf Hamiltonian whenever asigm.<sname>file is present.[gw].n1n2n3— GW k-mesh; smaller than[bz].nkabc(1/2 or 2/3).- LDA+U: add
idu / uh / jharrays inside[[spec]]; see the LDA+U section inlmf.md.
For the run-time -v syntax, see TOML migration:
# OLD (legacy ctrl + %const)
lmf si -vnk=8 -vmetal=3 -vnspin=2
# NEW (TOML path; processed in-memory, no on-disk rewrite)
lmf si --ctrlg:bz.nkabc=[8,8,8] --ctrlg:bz.metal=3 --ctrlg:ham.nspin=2⚠️ CAUTION —
-vused to override a%const NAME=...symbol declared inside the legacyctrl.<sname>({NAME}then expanded wherever it was referenced). Under TOML,-vpoints directly at a TOML path (--ctrlg:section.key=val); there is no%constindirection. So habits like-vmetal=3silently do nothing — use--ctrlg:bz.metal=3. See the CAUTION block on toml_migration.
lmchk mp-2534 --ctrlg:verbose=60allows you to check the recognized symmetries. (Legacylmchk --pr60 mp-2534is retired — the--pr=Nshortcut now aborts with a migration hint; use--ctrlg:verbose=Ninstead.)
At this point, you can visually check:
SiteInfo.lmchk— MT radii, atomic positionsPlatQlat.chk— primitive lattice vectors (plat) and reciprocal (qlat)
Detailed reference for every key in ctrlg.<sname>.toml.
ctrlgenToml.py↔ctrlgenM1.py(legacy) —ctrlgenToml.pyshares the periodic-table defaults withctrlgenM1.py(atomlist extracted at runtime), so both produce equivalent physics. The legacy two-step pathctrlgenM1.py <sname> → ctrlgenM1.ctrl.<sname> → cp to ctrl.<sname> → Legacy2toml.py <sname>is preserved for migrating old directories that already have hand-editedctrl.<sname>(orctrl.<sname>+GWinput) decks. See Step 2-Migration below.
Step 2-Migration. Existing ctrl.<sname> (and optional GWinput)
If you start from an old working directory that already has a hand-edited ctrl.<sname> (and possibly GWinput), don't rebuild from ctrls; convert in place:
Legacy2toml.py mp-2534This walks ctrl.<sname> and (if present) GWinput and emits ctrlg.<sname>.toml + PB.<sname>.toml. It is idempotent and prints [INFO] / [WARN] / [ERROR] diagnostics for any %const / -v overrides that won't survive as-is. See TOML migration for the full key map.
Install VESTA
It is convenient to see crystal structures with VESTA. (I installed VESTA-gtk3.tar.bz2 (ver. 3.5.8, built on Aug 11 2022, 23.8MB) on ubuntu 24) At ecalj/StructureTool/, we have 'viewvesta' command. Try
viewvesta ctrlg.si.toml # canonical (post-2026-05)
viewvesta ctrls.si # legacy structure-only seed fileto see the structure in VESTA. The converters vasp2ctrl / ctrl2vasp (also in /StructureTool) accept the same TOML / ctrls / ctrl input forms. (We have ~/ecalj/GetSyml/README.org. but Users do not need to read this.)
Step 3. LDA calculation
Run lmfa at first. It is for spherical atomic electron densities, contained in the crystals. lmfa ends instantaneously.
bashlmfa mp-2534gives spherical atom calculation for initialization.
lmfareadsctrlg.mp-2534.toml(auto-detected if omitted, when there is exactly onectrlg.*.tomlin the cwd) and generates the files required forlmfbelow. Checkconfsection in the console output asbashlmfa mp-2534 |grep conf. This shows atomic configuration (there are no side effects even if you run
lmfarepeatedly) asconf:------------------------------------------------------ conf:SPEC_ATOM= Ga : --- Table for atomic configuration --- (n and nz are Pr. Quantum numbers for valence) conf: isp l n nz Qval Qcore CoreConf conf: 1 0 4 0 2.000 6.000 => 1,2,3, conf: 1 1 4 0 1.000 12.000 => 2,3, conf: 1 2 4 3 10.000 0.000 => conf: 1 3 4 0 0.000 0.000 => conf: 1 4 5 0 0.000 0.000 => conf: Species Ga Z= 31.00 Qc= 18.000 R= 2.230000 Q= 0.000000 nsp= 1 mom= 0.000000 conf: rmt rmax a= 2.230000 49.011231 0.015000 nrmt nr= 661 867 conf: Core rhoc(rmt)= 0.001940 spillout= 0.001839 conf:------------------------------------------------------ conf:SPEC_ATOM= As : --- Table for atomic configuration --- (n and nz are Pr. Quantum numbers for valence) conf: isp l n nz Qval Qcore CoreConf conf: 1 0 4 0 2.000 6.000 => 1,2,3, conf: 1 1 4 0 3.000 12.000 => 2,3, conf: 1 2 4 0 0.000 10.000 => 3, conf: 1 3 4 0 0.000 0.000 => conf: 1 4 5 0 0.000 0.000 => conf: Species As Z= 33.00 Qc= 28.000 R= 2.330000 Q= 0.000000 nsp= 1 mom= 0.000000 conf: rmt rmax a= 2.330000 49.695135 0.015000 nrmt nr= 675 879 conf: Core rhoc(rmt)= 0.011286 spillout= 0.017467With this table, you can see the number of valence electrons of initial condition for Ga are 2,1, and 10 for 4s,4p, 4d+3d(with local orbital). Core configulation and charges are shown as well. Q=0 means satisfying charge neutrality.
- lmfa calculates spherical atoms (virtually ), and calculate logarithmic derivatives at MT boundaries, stored into
atmpnu.*. - The initial electron density for lmf is given as the superposition of the spherically symmetric atomic densities given by lmfa.
- When
[ham].readp = true(set byctrlgenToml.pyby default; legacyREADP=Twas the same), we keep the logarithmic derivative of the radial wave function at MT boundaries fixed.
- lmfa calculates spherical atoms (virtually ), and calculate logarithmic derivatives at MT boundaries, stored into
After
lmfa, we run LDA calculation as:bashmpirun -np 8 lmf mp-2534It will finish with several seconds. Then we see
bashit 10 of 80 ehf= -8398.499465 ehk= -8398.499466 From last iter ehf= -8398.499464 ehk= -8398.499465 diffe(q)= -0.000001 (0.000004) tol= 0.000010 (0.000010) more=F c ehf(eV)=-114268.304015 ehk(eV)=-114268.304037 sev(eV)Here
cis the sign thatdiffe(q),which shows the changes of energy and charge from the previous iteration, is less than the criterion, that is, converged. save.mp-2534 contains a line per iteration. We see it takes 10 times to have convergence. We show two energies, which should be the same.- Note: mp-2534 (GaAs) gives 5.75 for GaAs, while the experimental value is 5.65. I guess this is a PBEsol problem.
- llmf contains information of iterations (since I use tee command above), check eigenvalue and fermi energies, band gap.
- rst.mp-2534 is generated. Self-consistent charge included.
- You can change lattice constant by editing
[struc].alatinctrlg.mp-2534.toml. Math operators in TOML scalars are not evaluated; precompute the value (e.g.alat = 10.6818) or use--ctrlg:struc.alat=10.6818at run-time. The legacyctrl.<sname>%constmath (1.8897268777743552*5.65/5.75etc.) was baked in atLegacy2toml.pytime. - Note (historical):
ctrlp.<sname>was an intermediate file generated byctrl2ctrlp.pyfrom the legacyctrl.<sname>. As of 2026-05 the Fortran readsctrlg.<sname>.tomldirectly viam_ctrl_toml_loader.f90; noctrlpstep occurs. - check save.mp-2534. Show history of lmfa and lmf. one line per iteration. Show your console options. c,x,i,h LDA energy shown two values need to be the same (but slight difference). Repeat lmf stops with two iteration.
- SiteInfo.lmchk : Site infor
- PlatQlat.chk : Lattice info
- efermi.lmf: the Fermi energy
- estaticpot.dat : electrostatic potential of smooth part.
- __mixm* is the mixing file containing history of iteration.
- If you restart lmf again, it automatically read rst file. Thus it should be already converged-- to make it sure, it stops after two iterations.
Be careful about the definition of zero level of the one-body potential. We set the average of electrostatic potential at MT boundaries are zero.__*are temporary files, which can be deleted. As for the console out put, you see
...
bndfp: kpt 1 of 29 k= 0.0000 0.0000 0.0000 ndimh = nmto+napw = 82 55 27
bndfp: kpt 2 of 29 k= 0.0355 -0.0126 0.0000 ndimh = nmto+napw = 79 55 24
bndfp: kpt 3 of 29 k= 0.0710 -0.0251 0.0000 ndimh = nmto+napw = 82 55 27
bndfp: kpt 4 of 29 k= 0.1065 -0.0377 0.0000 ndimh = nmto+napw = 83 55 28
bndfp: kpt 5 of 29 k= 0.1420 -0.0502 0.0000 ndimh = nmto+napw = 89 55 34
bndfp: kpt 6 of 29 k= 0.0355 0.0251 0.0000 ndimh = nmto+napw = 78 55 23
bndfp: kpt 7 of 29 k= 0.0710 0.0126 0.0000 ndimh = nmto+napw = 80 55 25
..., where we see the Hamiltonian dimensions. When PWMODE=11, we have q-dependent energy cutoff of APWs by |q+G|^2< pwemax. (In our code, we use Rydberg ).
NOTE: In defaults given by ctrlgenM1.py, all the calculation are by
No empty spheres. EH=-1,EH=-2, MT radius is -3% untouching. RSMH=RSMH2=R/2
We do not claim this setting is the best, however, non-linear optimization is very problematic. We do not like to do one by one optimizaiton. Changing this setting might require extensive systematic study.
job_tdos, job_pdos
You can calculate total dos by
job_tdos mp-2534 -np 8
gnuplot -persist tdos.mp-2534.gltand pdos by
job_pdos mp-2534 -np 8job_pdos is a little expensive because symmetry is not used. After finished, gnuplot -p pdos.site001.mp-2534.glt shows the PDOS resolved into channels of real harmonics (shown at the end of job_pdos). 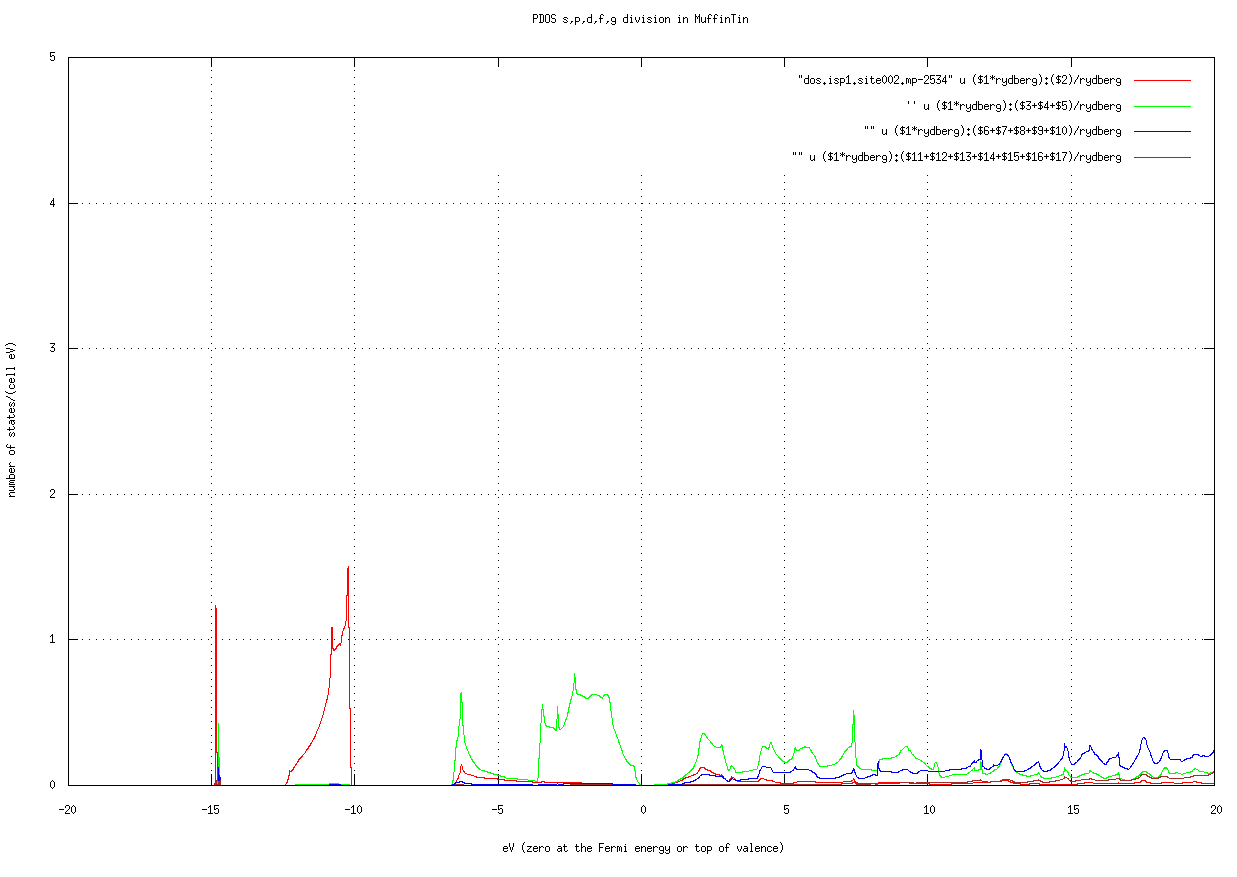 .This is PDOS at As site of mp-2534. Be careful about the number of k points. You may need to enlarge the mesh for accurate DOS / PDOS. Pass it on the command line with the TOML-path syntax:
.This is PDOS at As site of mp-2534. Be careful about the number of k points. You may need to enlarge the mesh for accurate DOS / PDOS. Pass it on the command line with the TOML-path syntax: job_tdos mp-2534 -np 8 --ctrlg:bz.nkabc=[10,10,10]. Check by grep '^bz.nkabc\|^nkabc' llmf_tdos and look at save.mp-2534.
Step 4. Create k-path and Brillouin zone for band plot
After lmf converges, make a band plot with job_band explained later on. The normality of the calculation of bands can be confirmed by the band plot (for magnetic systems, check the total magnetic moment and the magnetic moment for each site).
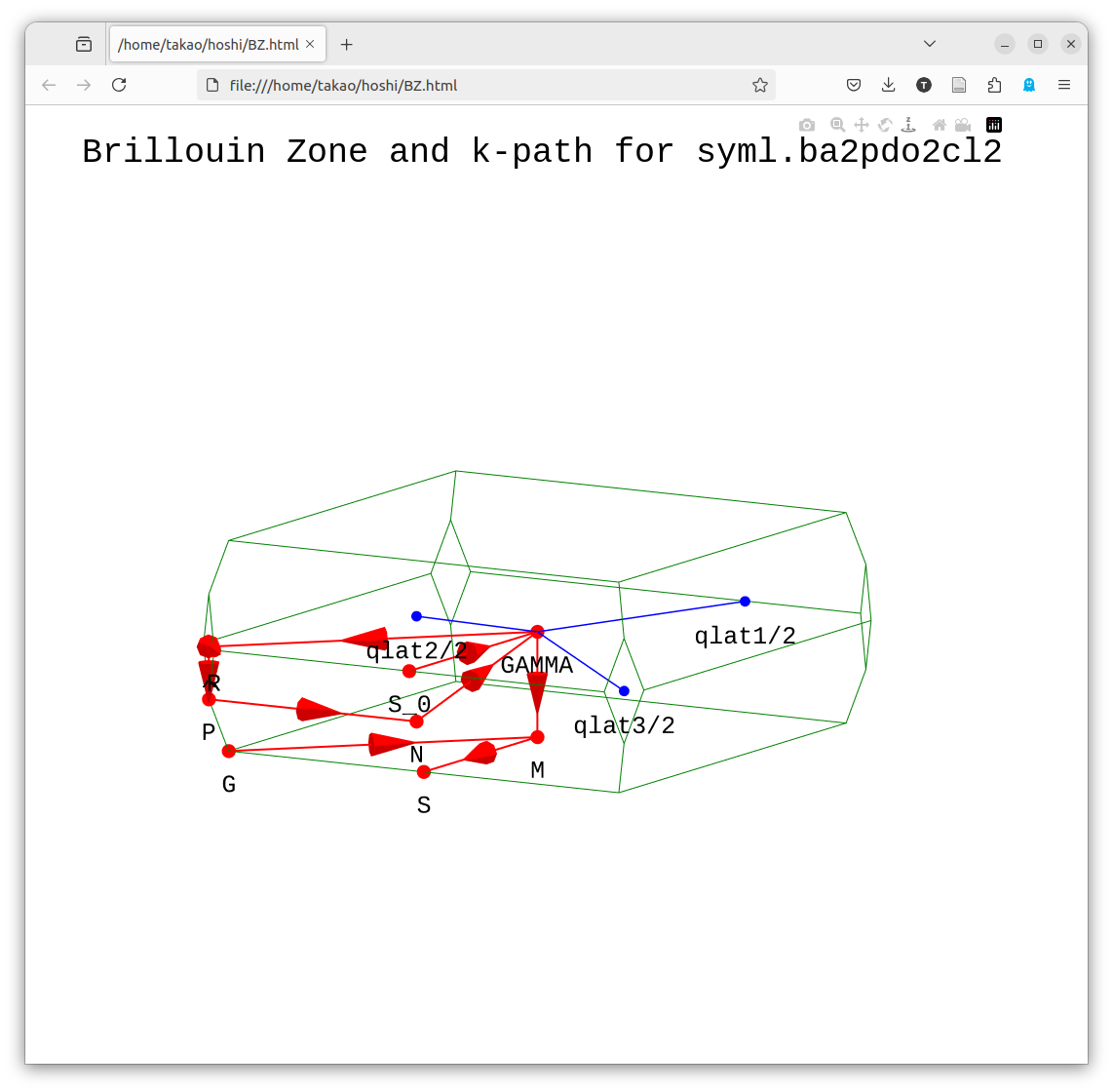
Before job_band, run getsyml gaas. Install any missing packages with pip. It is on spglib by Togo and seekpath. After finished, view BZ.html. It shows the k-path in the BZ. We show an example for ba2pdo2cl2 in the figure below. It is an interactive figure written with plotly, so you can read the coordinate values.
getsyml foobar(foobar here matches the <sname> in ctrlg.<sname>.toml.)
- Samples of BZ.html by getsyml are seen at https://ecalj.sakura.ne.jp/BZgetsyml/
Step 5. band plot
(this is a case for ba2pdo2cl2 )
>job_band ctrl.ba2pdo2cl2 -np 8A gnuplot script can be created. Edit it if necessary. If you edit syml.ba2pdo2cl2 before job_band, you can adjust the symmetry line and mesh size.
- The following picture is the LDA bands for the default calculation of ba2pdo2cl2 (the names of the symmetric points can be confirmed with BZ.html. In addition, look into
syml.<sname>). 0 eV is the Fermi energy. Since this is metallic, we see no band gap.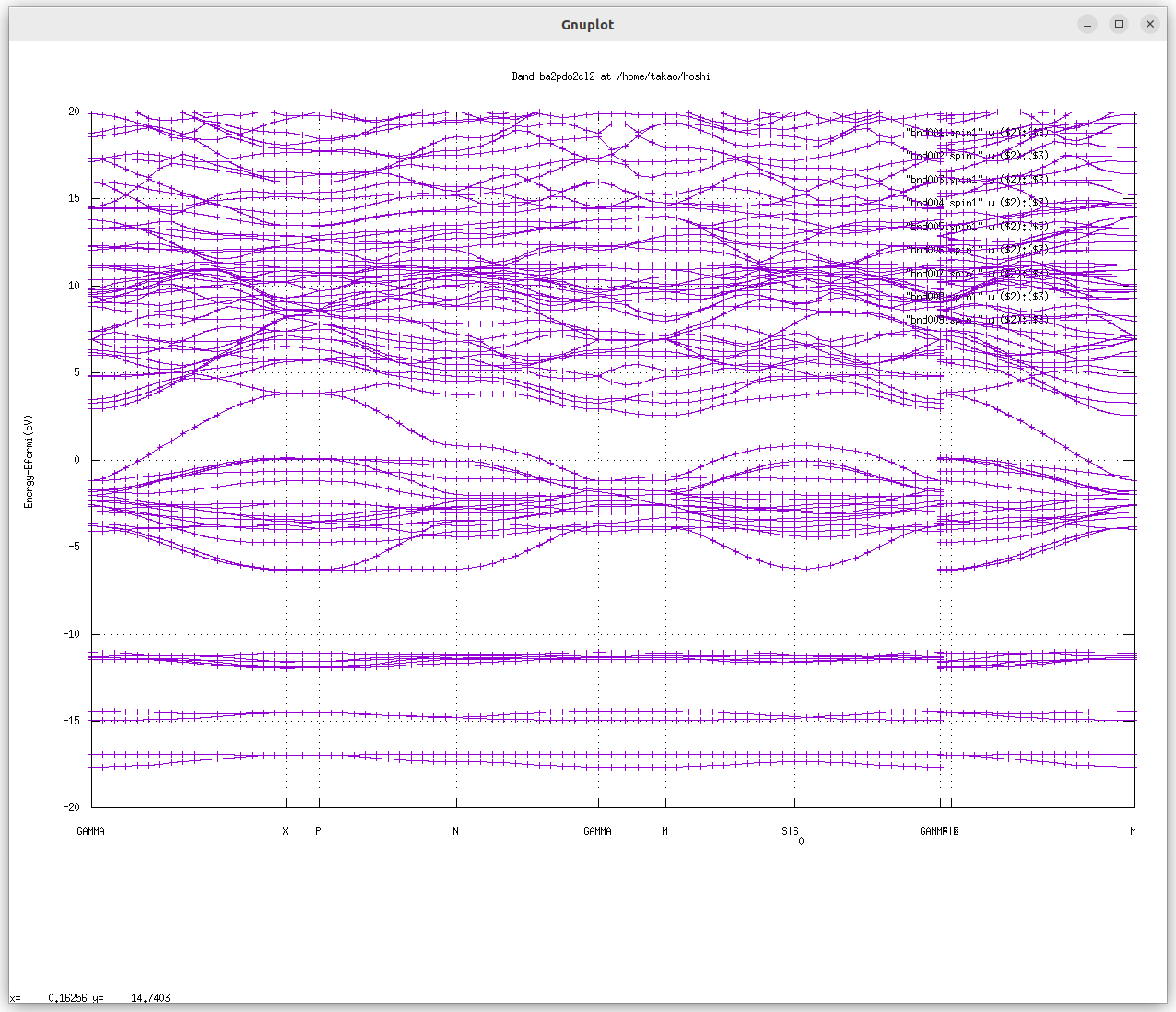 .
.
Here we are talking about band energies.
In ecalj, the k mesh for
lmf([bz].nkabcinctrlg.<sname>.toml) and the k mesh for GW ([gw].n1n2n3) can be different. The former affects LDA wall-time only weakly; the latter has a large effect (so we want to reduce[gw].n1n2n3).In ecalj's band plot mode, theoretically degenerated bands because of symmetry at the BZ edge are not degenerated. This is because there are limited numbers of APW basis functions, so run the band plot with pwemax=4, etc. (Temporary solution: We want to automate it).
job_tdos, job_fermisurface, job_pdos
job_pdos calculates PDOS, job_tdos calculates total DOS, and job_fermisurface draws the Fermi surface with Xcrysden. job_fermisurface can be used to draw the shape of the CBM bottom as ellipsoid of Si. xxx
Step 6. QSGW calculation
We now run QSGW calculations. QSGW is computationally very expensive, so we recommend running smaller systems first.
Since 2026-05 the GW driver inputs are inside the same
ctrlg.<sname>.toml, in sections[gw],[product_basis], and[blocks]. The legacy separateGWinputtext file is no longer parsed by Fortran. If you started from an old directory withctrl.<sname>+GWinput,Legacy2toml.py <sname>(Step 2.5) already merged the GW content for you. If you have onlyctrl.<sname>(noGWinput) and want a default GW set-up, run:bashmkGWinput mp-2534 # produces a legacy GWinput template Legacy2toml.py mp-2534 # re-emit ctrlg.mp-2534.toml with [gw] etc.
The single key you most often tweak before launching QSGW is the GW k-mesh, [gw].n1n2n3, kept smaller than [bz].nkabc (typically 1/2 or 2/3 of it) since it dominates wall-time:
[bz]
nkabc = [12, 12, 12] # LDA k-mesh
[gw]
n1n2n3 = [6, 6, 6] # GW k-mesh (smaller -> faster)For Si, n1n2n3 = [6, 6, 6] is good; the rest of [gw] rarely needs touching for non-magnetic semiconductors. The legacy GWinput key mapping lives in lmf.md § Legacy ctrl.<sname> ↔ TOML path map, and the historical GWinput text format is documented (as a reference only) in gwinput.md. Here is a convergence behavior of the band gap for GaAs taken from Ref.[3], 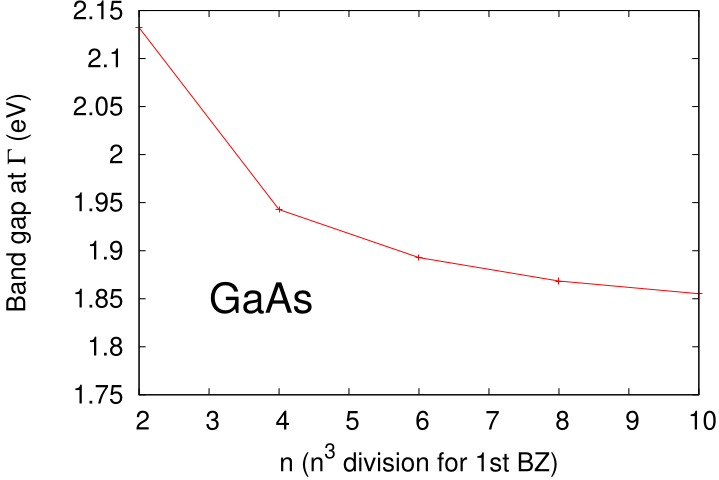 From this picture, I may say 4 4 4 is not so bad if we assume ~0.1eV accuracy. 6 6 6 is good. More than 8 8 8 is for numerical check(not fruitful for practical applications).
From this picture, I may say 4 4 4 is not so bad if we assume ~0.1eV accuracy. 6 6 6 is good. More than 8 8 8 is for numerical check(not fruitful for practical applications).
Flow of QSGW calculation with the script gwsc
We run the QSGW calculations with gwsc. For semiconductors, several QSGW iterations are fine, close enough to final results. QSGW is to obtain band structures (or one-body Hamiltonian), the total energy is not yet.
QPU file contains diagonal components of GW calculations. Note that our Mixed Produce basis is a key technology for the GW calculation.
gwsc -np NP [--gpu] [--mp] nloop extension(For magnetic materials wanting the same basis for up and down, set [ham] phispinsym = true in ctrlg.<sname>.toml, or pass --ctrlg:ham.phispinsym=true at run time.)
Then console outputs of gwsc is somthing like
### START gwsc: ITERADD= 1, MPI size= 4, 4 TARGET= si
===== Ititial band structure ======
---> No sigm. LDA caculation for eigenfunctions
0:00:00.226245 mpirun -np 1 /home/takao/bin/lmfa si >llmfa
0:00:00.807062 mpirun -np 4 /home/takao/bin/lmf si >llmf_lda
===== QSGW iteration start iter 1 ===
0:00:03.071054 mpirun -np 1 /home/takao/bin/lmf si --jobgw=0 >llmfgw00
0:00:03.904403 mpirun -np 1 /home/takao/bin/qg4gw --job=1 > lqg4gw
0:00:04.431022 mpirun -np 4 /home/takao/bin/lmf si --jobgw=1 >llmfgw01
0:00:05.918216 mpirun -np 1 /home/takao/bin/heftet --job=1 > leftet
0:00:06.444439 mpirun -np 1 /home/takao/bin/hbasfp0 --job=3 >lbasC
0:00:07.064558 mpirun -np 4 /home/takao/bin/hvccfp0 --job=3 > lvccC
0:00:07.812283 mpirun -np 4 /home/takao/bin/hsfp0_sc --job=3 >lsxC
0:00:08.545956 mpirun -np 1 /home/takao/bin/hbasfp0 --job=0 > lbas
0:00:09.156775 mpirun -np 4 /home/takao/bin/hvccfp0 --job=0 > lvcc
0:00:09.884064 mpirun -np 4 /home/takao/bin/hsfp0_sc --job=1 >lsx
0:00:10.644292 mpirun -np 4 /home/takao/bin/hrcxq > lrcxq
0:00:11.482931 mpirun -np 4 /home/takao/bin/hsfp0_sc --job=2 > lsc
0:00:12.460776 mpirun -np 1 /home/takao/bin/hqpe_sc > lqpe
0:00:13.019735 mpirun -np 4 /home/takao/bin/lmf si >llmf
===== QSGW iteration end iter 1 ===
OK! ==== All calclation finished for gwsc ====...
The console outputs are redirected to log files l*. lsxC is the exchange self-energy due to cores. lsx is for exchange. lsc is correlation. lvcc is for Coulomb matrix。 In this calculation we run gwsc -np 8 1 si, where 1 is the number of QSGW iteration. If you repeat gwsc, we have additional QSGW iterations on top the previous calculations.
a case of La2CuO4
For La2CuO4, I had (verbatim log from 2025-06; the legacy -vssig=0.8 form recorded below is no longer parsed — today the same override is --ctrlg:ham.scaledsigma=0.8):
2025-06-27 19:09:01.465241 mpirun -np 1 echo --- Start gwsc ---
--- Start gwsc ---
option= -vssig=0.8
### START gwsc: ITERADD= 5, MPI size= 32, 32 TARGET= lcuo
===== Ititial band structure ======
---> We use existing sigm file
0:00:00.041902 mpirun -np 32 /home/takao/bin/lmf lcuo -vssig=0.8 >llmf_start
We found QPU.5 -->start to generate QPU.6...
===== QSGW iteration start iter 6 ===
0:00:18.111042 mpirun -np 1 /home/takao/bin/lmf lcuo -vssig=0.8 --jobgw=0 >llmfgw00
0:00:18.233440 mpirun -np 1 /home/takao/bin/qg4gw -vssig=0.8 --job=1 > lqg4gw
0:00:18.488197 mpirun -np 32 /home/takao/bin/lmf lcuo -vssig=0.8 --jobgw=1 >llmfgw01
0:00:40.910973 mpirun -np 1 /home/takao/bin/heftet --job=1 -vssig=0.8 > leftet
0:00:41.052760 mpirun -np 1 /home/takao/bin/hbasfp0 --job=3 -vssig=0.8 >lbasC
0:00:43.290214 mpirun -np 32 /home/takao/bin/hvccfp0 --job=3 -vssig=0.8 > lvccC
0:01:00.327823 mpirun -np 32 /home/takao/bin/hsfp0_sc --job=3 -vssig=0.8 >lsxC
0:01:45.547149 mpirun -np 1 /home/takao/bin/hbasfp0 --job=0 -vssig=0.8 > lbas
0:01:46.806858 mpirun -np 32 /home/takao/bin/hvccfp0 --job=0 -vssig=0.8 > lvcc
0:01:59.034735 mpirun -np 32 /home/takao/bin/hsfp0_sc --job=1 -vssig=0.8 >lsx
0:02:45.526614 mpirun -np 32 /home/takao/bin/hrcxq -vssig=0.8 > lrcxq
0:06:51.996781 mpirun -np 32 /home/takao/bin/hsfp0_sc --job=2 -vssig=0.8 > lsc
0:23:31.022636 mpirun -np 1 /home/takao/bin/hqpe_sc -vssig=0.8 > lqpe
0:23:33.062303 mpirun -np 32 /home/takao/bin/lmf lcuo -vssig=0.8 >llmf
===== QSGW iteration end iter 6 ======== QSGW iteration start iter 7 ===
0:24:12.969096 mpirun -np 1 /home/takao/bin/lmf lcuo -vssig=0.8 --jobgw=0 >llmfgw00
...This is without GPU. We see one QSGW iteration requires 24 minutes (start timing is shown at the top of lines). Since I had 5th-QSGW iteration finished (checked by the existence of QPU.5run), it start from 6th iteration.
a case study of ba2pdo2cl2.
If you started from a pre-2026-05 directory with only ctrl.<sname>, generate the GW driver settings and merge them into TOML in two steps:
mkGWinput ba2pdo2cl2 # produces a legacy GWinput.tmp template
cp GWinput.tmp GWinput # copy and (optionally) edit
Legacy2toml.py ba2pdo2cl2 # ctrl.<sname> + GWinput -> ctrlg.<sname>.toml + PB.<sname>.tomlAfter conversion, the only key you usually need to tweak before running QSGW is the GW k-mesh:
[gw]
n1n2n3 = [4, 4, 4] # smaller than [bz].nkabc; 1/2 or 2/3 of it worksFor non-magnetic semiconductors the rest of [gw] rarely needs attention.
Then you can run QSGW calculation with
gwsc -np 32 1 ba2pdo2cl2. Here 1 means the number of QSGW iterations. QSGW iteration is quite time-consuming. gwsc gives minimum help (we need to explain options elsewhere).
The iteration is kept in rst.<sname>:electron density, sigm.*:vxcqsgw. (Remove these files in addition to *run files/directories if you like to start from the beginning).
It requires 53 minutes to run one iteration of QSGW.
job_band ba2pdo2cl2 -np 32 gives the following picture. QSGW one-shot changes band structure around Ef from that in LDA.But still metallic, no band gaps.
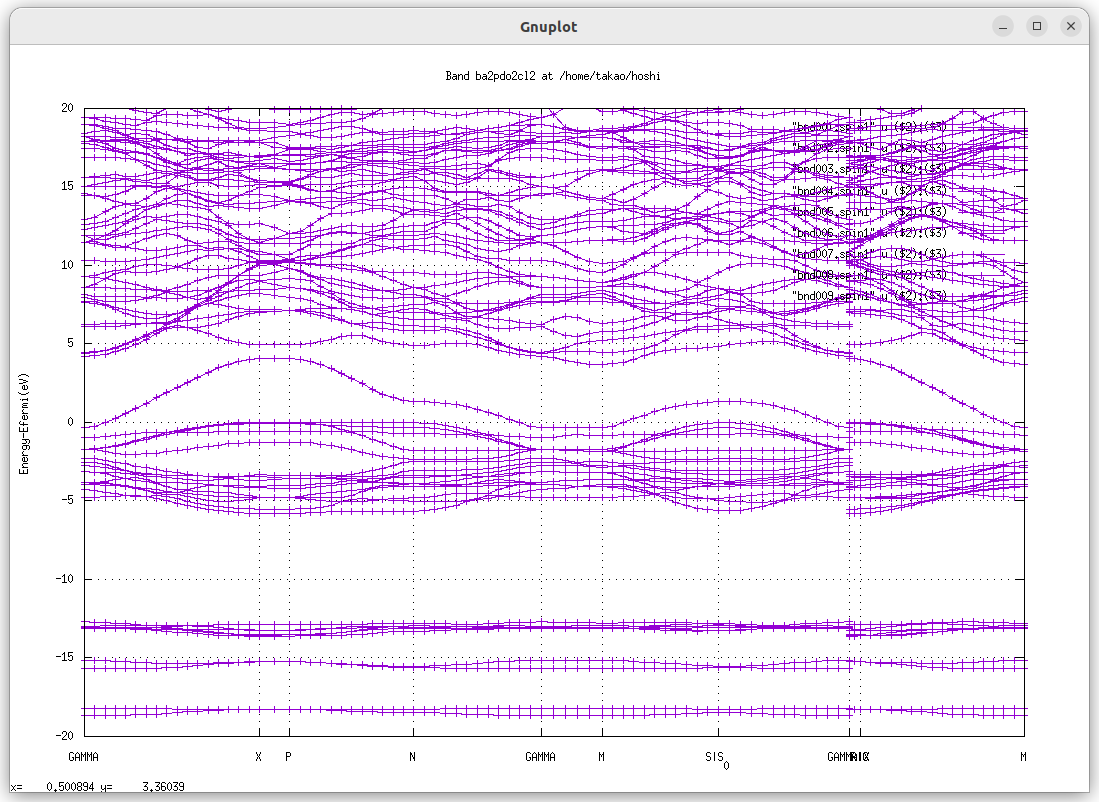
To continue QSGW iteration, run
gwsc -np 32 nx ba2pdo2cl2Since you did 1 already. You will have the results of 1+nx QSGQ iteration.
when we run 8 iterations as for ba2pdo2cl2, we had band gap 2.1 eV. We saw band gap after 4th iteration.
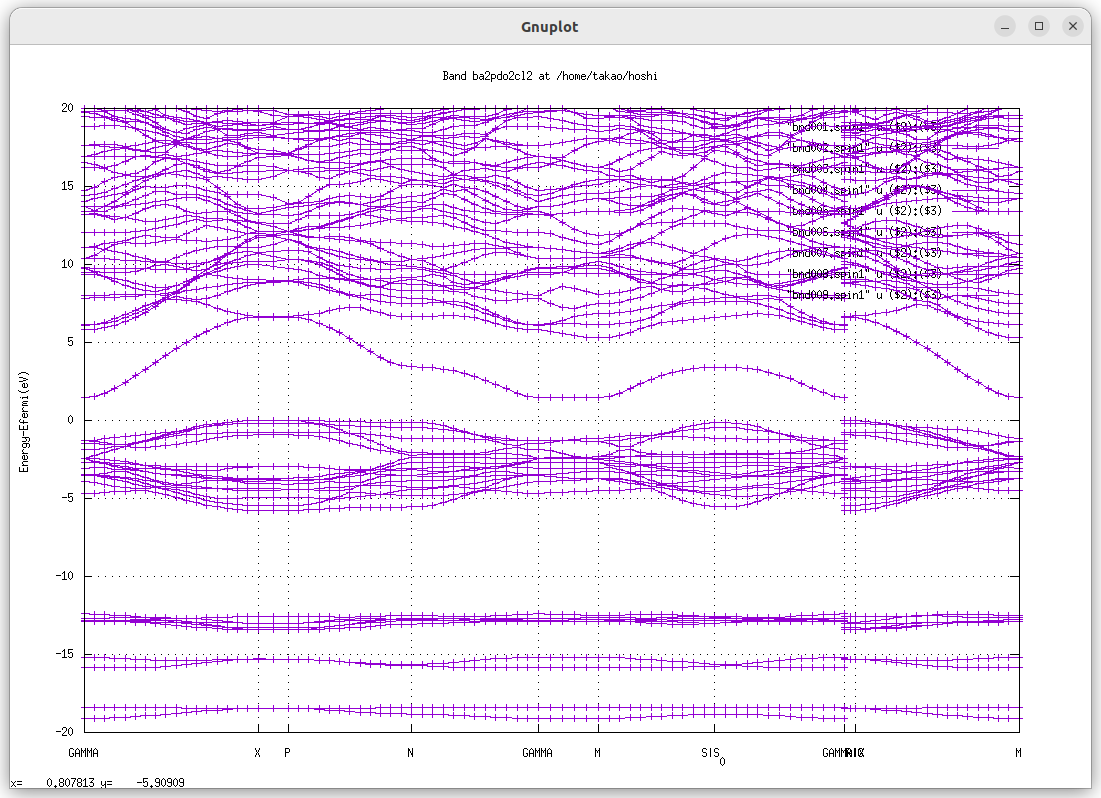
For comparison with experiments, we recommend to use ssig=0.8 (set in
ctrlg.<sname>.tomlas[ham].scaledsigma = 0.8), which is called as QSGW80.Spin-orbit coupling. After you obtain
sigm.<sname>, you set SO=1 (LdotS scheme) and run lmf. Then you can include effect of SOC. Sinc SO=1 is not implemented in the whole gwsc cycle, we have to include SOC just at the end step (We include SOC after we fix VxcQSGW).If you run
gwsc -np 32 5 ba2pdo2cl2 -vssig=0.8, this overide ssig, which is defined in ctrl.ba2pdo2cl2, in lmf calculations. (Check it in save.ba2pdo2cl2)
a case of KTaO3
Example of QSGW for KTaO3 (perovskite,mp-3614) 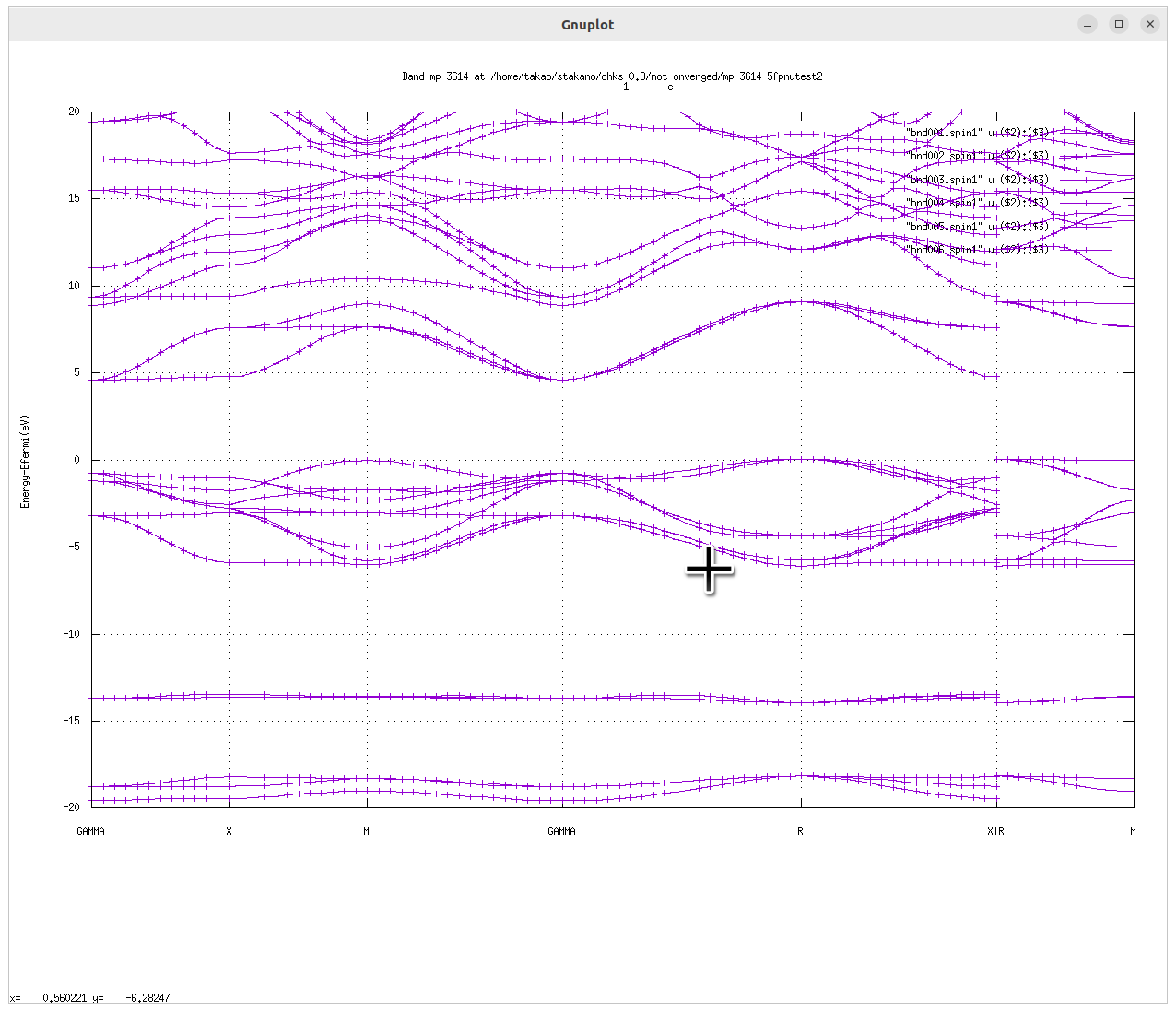
lmchk
lmchk mp-2534is to check the crystal symmetry. In addition determine MT radius. and Check the ovarlap of MTs. Defaults setting is with -3% overlap.(no overlap).
- symmetry
- MT overlap If you have less symmetry rather than the symmetry of lattice for magnetic systems, you have to set crystal symmetry by hand.
This can be done by adding space group symmetry generator to SYMGRP (instead of find). We need to pay attention for this point in the case of SOC.
how to write space-group operation
For example r3x means 3-fold axis along x. How to express space-group operations in ecalj are explained at SYMGRP section at lmf.md.
How to start over calcualtions
Remove mix* rst* (mix* is mixing files) If MT radius are changed, start over from lmfa (remove atm* files)
- As long as converged, no problem.
- If you have 3d spagetti-like entangled bands at Ef, need caution.
jobmaterials.py: mini database for computational tests
At ecalj/MATERIALS/, type ./jobmaterials.py. It shows a help with a list of materials. It contains samples of simple materials. It performs LDA calculations and generates GW driver settings (now in [gw] of ctrlg.<sname>.toml; legacy GWinput for un-migrated materials). (I think MATERIALS/ is not organaized well. We are going to clean up)
- How to rungives a help, showing a list of materials as
./job_materials.py=== Materials in Materials.ctrls.database are:=== 2hSiC 3cSiC 4hSiC AlAs AlN AlNzb AlP AlSb Bi2Te3 C CdO CdS CdSe CdTe Ce Cu Fe GaAs GaAs_so GaN GaNzb GaP GaSb Ge HfO2 HgO HgS HgSe HgTe InAs InN InNzb InP InSb LaGaO3 Li MgO MgS MgSe MgTe Ni NiO PbS PbTe Si SiO2c Sn SrTiO3 SrVO3 YMn2 ZnO ZnS ZnSe ZnTe ZrO2 wCdS wZnS
Run Si for example:
./job_materials.py Siperforms LDA calculation of Si at ecalj/MATERIALS/Si/. '--all' works as well instead of 'Si'.
job_materials.py works as follows for given names. Step 1. Generate ctrls.* file for Materials.ctrls.database. (names are in DATASECTION:) Step 2. Generate
ctrlg.<sname>.toml+PB.<sname>.tomlbyctrlgenToml.py(legacy path:ctrlgenM1.py+Legacy2toml.py) Step 3. Make directory such as Si/ and copy ctrls.si plus the generated TOML pair (legacy: ctrls.si, ctrl.si, GWinput) Step 4. run lmfa and lmf We can skip step 4 with--noexec. Run "job_materials --all --noexec" is an idea to generate all input files for materials in the database. Watch no strange erros appear.
Key input files are
ctrls.si,ctrl.si. See sections below.rst.sicontains self-consistent electron density. Check iterations with the output filesave.si. The console output of lmf is in llmf. Not need to know all the console outputs.Before QSGW, it is better to confirm the LDA level calculations are fine. In order to do the confirmation, band plot is convenient. For band plot we need the symmetry line as
syml.siwhich can be generated bygetsyml siThen run
job_band si -np 8results band plots in the gnuplot.
This is the end of GetStarted. Goto UsageDetailed